Block 2 - COOIII: Horizontal gene transfer in a gene tree of an aminoacyl-tRNA synthetase
Overview
Teaching: 0.0 min
Exercises: 60 minQuestions
Can we recognize HGT in a gene tree?
Objectives
Practice with making trees
Practice with recognizing HGT?
Horizontal gene transfer and the COG database
We are going to reproduce a (classic) finding on horizontal gene transfer from the literature. We will use the COG database which is a collection of (inclusive) Clusters of Orthologous Genes (hence COG) that have also been annotated with a function common to all genes in a COG, as well as solving common (and ancient) gene fusions and fissions. The COGs are frequently used for genome annoations in prokaryotes they represent decent orthogroups and have function attached to them. For teaching and looking at trees they are also convenient because the original COG annoation was based on a limited number of species, allowing us to make trees and look at these trees.
setting up your environment and getting some data from the COG database
Get to gemini and get prepared for this COO (used from dr. D. Tamarit)
$ cd ~/GenomeBioinformatics/Block2/
$ mkdir COOIII
$ cd COOIII
We are going download from the the COG database the file that contains a list of all the COGs, their functions and the proteins assigned to each COG. In the public ftp folder https://ftp.ncbi.nlm.nih.gov/pub/COG/COG/, it is the file labeled whog. You have multiple options to do this. For example you can use wget to directly get this file into your COOIII folder in the following way
$ wget https://ftp.ncbi.nlm.nih.gov/pub/COG/COG/whog
Alternatively, you could falso go the ftp site (https://ftp.ncbi.nlm.nih.gov/pub/COG/COG/), download the whog file to your laptop, and from your laptop use scp to copy the whog file to gemini.
No matter how you got the file, inspect the file and use e.g. grep or some other trick to count the number of COGs in the file.
Exercise: How many COGs does the whog file contain?
Solution
by doing
grep -c COG whogor control-F in some text editor
you should find that the file contains 4873 COGs
Getting all the sequences belonging to COG0072 into a fasta file
Search for COG0072 in this “whog” file and inspect it.
exercises: What is its function of COG0072
Solution
[J] COG0072 Phenylalanyl-tRNA synthetase beta subunit
so a subunit of the t-RNA synthetases for phenylalanyl (F) that couples the tRNA(s) for this amino acid to that amino acid
Make a list file that contains all members of COG0072 such that we can use seqtk. (i.e. a simple text file with fasta sequence identifiers without the “>” sign below each other). comment: <> ( give them a script or regular expression to do this, rather than having them do it in a text editor )
exercises: How many identifiers do you have in your list file? (i.e. how many proteins where assigned in the whog file to COG0072
Solution
If you take the multiple identifiers for the eukaryotes into account: 68, otherwise a little bit less
The sequences that correspond to the identifiers from the whog file are present in ~/data_bb3bcg20/Block2/COOIII/myva.fa. Collect all sequences assigned to COG0072 from the database myva.fa using e.g. seqtk subseq ~/data_bb3bcg20/Block2/COOIII/myva.fa your_file_with_sequence_identifiers > your_new_fast_file.
exercises: How many sequences do you have in your fasta file?
Solution
Should be 68 sequences
Making and interpreting the gene tree of COG0072
Align your sequences using MAFFT (see previous COO’s for instructions)
Make a tree of these sequences using IQTREE. However, note that this is already a fairly large collection of sequences. So, I would advise to run IQTREE using the fast option using e.g. iqtree -s [your alignment file] -fast -m LG+G4. In this particular case for the bits of tree that we are interested in, running IQTREE with this fast option should not matter, but generally trees reconstructed using the fast option are much poorer. Feel free to run the tree with and without the fast option to compare.
Open your tree in iToL (see previous computer exercises for specific instructions on how to upload treefiles to iToL and visualize them there). Put the tree in a layout that you find most easy to browse.
To make sense of the tree it is relevant to know Ta, TVN, PH, PAB, PAE, SSO, APE, VNG, MJ, MTH, MK, MA, AF are all archaea; YLR060w, YPR047w_2, SPAC23A1.12c, SPCC736.03c_2, ECU04g0900 are eukaryotic sequences; and all other sequences are bacteria.
exercises: In this tree, can you annoate the speciations in this tree that more or less correspond to LUCA and potentially to LECA’s (potentially because the inference is uncertain given that the only eukaryotes are a few fungi) ?
Solution
See above, blue ball LUCA, red ball LECA
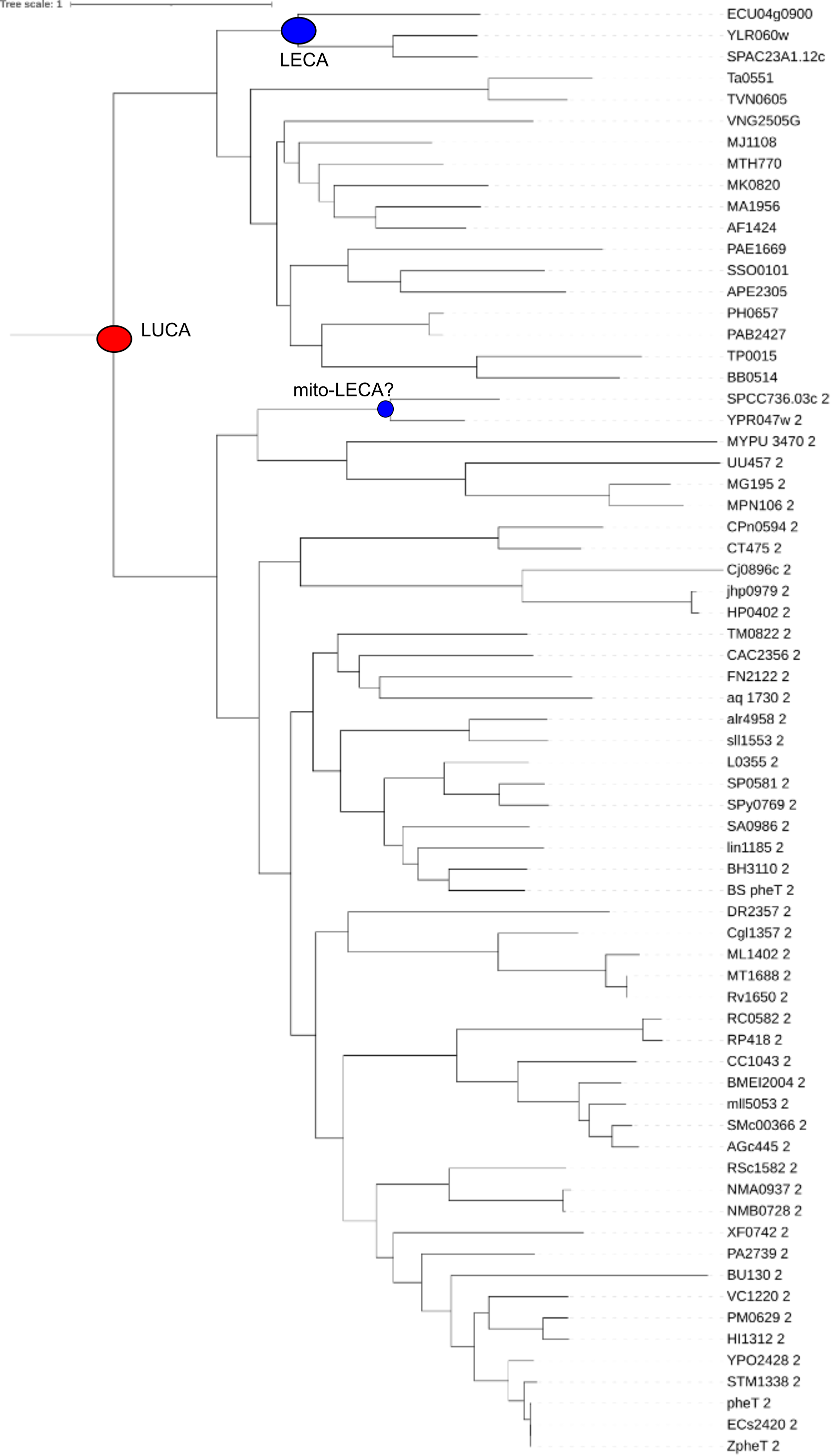
Try to find the inter domain HGT branch(es) in the tree. Also consider which genes are from the acceptor species and which lineage is the donor.
exercises: indicate in the tree where the HGT took place and who the acceptors and donors were
Solution
A colored tree , and with branches rotates to better isolate the transfer receivers, makes it easier to see perhaps … red, archaea, blue bacteria
transfer branch green
transfered genes: TP0015, BB0514, lineage donor is archaea
Use https://ftp.ncbi.nlm.nih.gov/pub/COG/COG/org.txt and https://ftp.ncbi.nlm.nih.gov/pub/COG/COG/whog to piece together the name of the lineage that was the recipient of transfer? spirochaetes (or in the file org.txt Chlam-Spir) )

exercises: Can you find the paper that inspired us to make this exercise?
Solution
We utilized this ancient paper: https://genome.cshlp.org/content/9/8/689.long as a source, because then we could use a limited species set, i.e. a rather old version of the COGs to see the transfer. In addition the author of the COGs is also the author of this paper making it likely that the inference and the data match.
This is a more recent paper that documents the tRNA synthetases: https://link.springer.com/article/10.1007/s00239-016-9768-2
In this more recent study, quite some interkingdom transfers can be seen in addition the “expect” (asgard)archaeal origin and endosymbiotic origins:
Key Points
The acceptor taxon is clustered within the donor taxon