Block 2 - COOI: Gene tree intepretation & orthology of an important metabolic enzyme
Overview
Teaching: 0.0 min
Exercises: 45 minQuestions
How is my human gene related to genes in other model organisms and what does this tell me about orthologs
Objectives
Practice the usage of common command line tools to find homologs and build gene trees.
Practice with interpreting gene trees.
Set up your environment
As instructed in the first computer exercises of block1, get to your work environment and get prepared for this COO. i.e. do something like
$ cd GenomeBioinformatics/
$ mkdir -p Block2/COOI
$ cd Block2/COOI
getting a sequence
In this exercise, our goal is to infer the evolutionary history of a human protein starting from its sequence. This evolutionary history should reveal the orthologs in other species and the timing of the duplicates of our protein. We are going to use the human 6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase as a starting point. This bi-functional enzyme (hence the long name) is an enzyme that catalyzes formation as well as degradation of a significant allosteric regulator of glycolysis and gluconeogenesis: fructose-2,6-bisphosphate.
Go to http://www.uniprot.org/uniprot/Q16877 . Quickly scan the page. And see what kind of information uniprot has available and to what kind of databases uniprot cross references.
In the uniprot page, click on sequence & isoforms, and there click on download. You should see the sequence and the fasta header of this enzyme. If you do not know what the fasta format entails see https://en.wikipedia.org/wiki/FASTA_format
We need this sequence to be on gemini in order to look for homologous sequences in other species in order to make a gene tree.
One way to do this is to use wget while you are in your working folder. Copy the URL (i.e. the web address) of the fasta file from your browser and do wget [copied_url_of_your_protein_sequence]. When we are going to do blast, the instructions assume the protein sequence that is going to serve as query is called query.txt. So maybe you should use cp to copy the file you obtained via wget to a file called query.txt.
Another way to do this is to open a text editor on your local laptop (e.g. textedit, notepad++) , and copy the protein sequence of Q16877 into a text file. Save the protein sequence as a text file named “query.txt”. Then use scp to copy the text file to your gemini folder where we are doing these exercises, i.e. for example using scp [location_of_sequence_file/name_of_sequence_file] [your_studentnumber_here]@gemini.science.uu.nl:GenomeBioinformatics/Block2/COOI/
Check how your file looks on gemini by typing e.g. more query.txt, or less query.txt; (or more Q16877.fasta if you obtained the fasta file via wgetand did not yet copy it into query.txt)
running blast
Now we are going to run blast with the human 6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase as a starting point. We are going to do this via blastp -query query.txt -db ~/data_bb3bcg20/Block2/COOI/proteomes1.fa > tmp.out
Look at the output, using (for example e.g. more tmp.out or less tmp.out). Proteins starting with HSAP are human. ATHA is a plant, CELE is a nematode worm, DMEL is the fruitfly, SCER is baker’s yeast, SPOM is fission yeast.
Exercise: How many hits with E-value < 1e-10 do you see in human, plant, worm and fly?
Solution
HSAP037297 979 0.0 HSAP082045 743 0.0 HSAP043809 671 0.0 HSAP095035 667 0.0 DMEL018546 529 0.0 CELE028867 477 3e-166 CELE024628 437 2e-150 SCER003727 406 2e-138 ATHA005880 345 1e-110 SPOM004734 304 4e-99 SCER001206 298 3e-92 SPOM002399 287 3e-89 SPOM003505 253 5e-77 SCER000727 202 1e-58 SPOM001690 166 2e-45So, HSAP 4, DMEL, 1, CELE 2, ATHA 1
Still looking at the blastoutput file, look at the pairwise alignment between your query and its best hit in DMEL.
Exercise: What is the percent identity with the best hit in fly?
solution
> DMEL018546
Length=716\
Score = 529 bits (1363), Expect = 0.0, Method: Compositional matrix adjust.
Identities = 258/456 (57%), Positives = 329/456 (72%), Gaps = 5/456 (1%)
so precent identity 57%
We want to create a fasta file in order to make a tree of the hits in plants, in animals (fly, worm, human) with E-value < 1e-10; and of the best hit in fission yeast (SPOM) and the best hit in baker’s yeast SCER. To do so:
Copy the identifiers of the sequences you want for the tree into a text file on your laptop, each identifier should be followed by a newline. Copy this file to gemini using scp (see instructions above for copying the fasta file). Then use seqtk subseq [fasta database] [name of your list of identifiers] > [your new file of homologous e.g. homs.fa] The fasta file you should use is the original database against which we performed our blast, e.g.~/data_bb3bcg20/Block2/COOI/proteomes1.fa.
Run mafft on your fasta file. i.e. mafft [yourfile e.g. homs.fa] > [name of alignment file, e.g. homs.msa]
Then run iq tree e.g. iqtree -s homs.msa –m LG+G4
Download the output tree (i.e. homs.msa.treefile) using scp to your laptop or perhaps easier, on the command line do cat homs.msa.treefile and copy the text from the screen to paste to view the tree in iToL https://itol.embl.de/upload.cgi
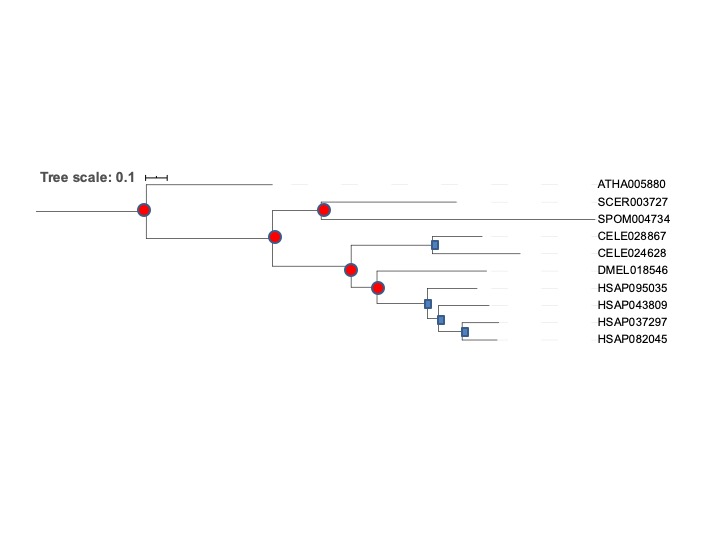
Okay now we get to the interpretation. Look at the tree and reroot it to make some kind of biological sense. You can reroot trees by clicking on a branch -> Tree structure -> re-root the tree here. Sketch the resulting tree on paper or copy a picture of the resulting tree into a program which allows to draw on top of it (e.g. powerpoint, paint, inkscape). Annotate the tree in terms of duplications and speciations.
Exercise: How many duplications does this tree imply?
solution
Rooted the tree on plants because officially fungi and animals are closer to eachother than either is to plants: So I get this
We have 4 duplications
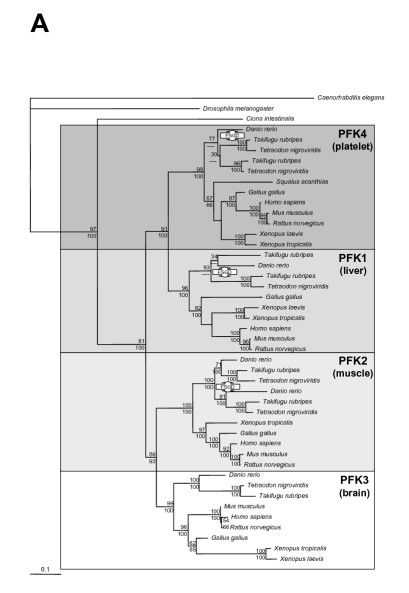
Check the function of the different human genes, and the reconstruction according to literature from the following article https://bmcbiol.biomedcentral.com/articles/10.1186/1741-7007-4-16 (our proteins are in the left most panel of figure 2).
Exercise: What type of functional differentiation have the genes undergone.
solution
Functional differentiation is change in tissue where these paralogous enzymes are predominantly expressed i.e. platelet, liver , muscle, brain. So no change in enzymatic/molecular function. This type of functional differentiation is common with inparalogs.
Go back to your tree and based on speciations and duplications consider orthology.
Exercise: According to your tree, which human gene(s) are orthologs of which gene(s) in D. melanogaster and to which gene(s) in C. elegans?
solution
HSAP037297, HSAP082045, HSAP043809, HSAP095035 are orthologous to DMEL018546 . 1-to-many.
HSAP037297, HSAP082045, HSAP043809, HSAP095035 are orthologous to CELE028867, CELE024628 Many-to-many
Key Points
Orthology can be a many-to-many relation